Anomalías Congénitas: 3 tipos
1) Anomalías de cantidad
*Agenesia: ausencia de riñón, si es bilateral es incompatible con la vida, unilateral, el otro riñon compensala función.
*Hipoplasia: menor desarrollo, si es bilateral lleva a insuficiencia en la infancia, unilateral, compensa el otro.
*supernumerarios
2)Anomalías de posición
*Ectopía: el riñon puede hallarse dentro de la pelvis con uréteres tortuosos, que dificultan la salida de orina, predispone a infecciones.
*Riñon en herradura: se fusionan ambos riñones por uno de sus polos, generalmente por el inferior.
3) Anomalías de diferenciación:
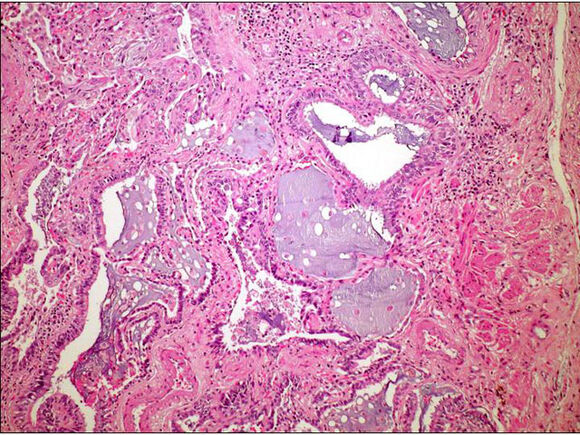
* Displasia quística renal: aumento del tamaño renal, si es bilateral lleva a la insuficiencia. Hay anomalías de la diferenciación, múltiples quistes con islotes de mesénquima intermedio (patognomónico). Hay nefronas normales junto a nefronas inmaduras. Los quistes se recubren de epitelio plano y contienen líquido seroso.
* Enfermedad Poliquística Renal:
-del adulto: siempre es bilateral, riñones poliquisticos que borran los detalles normales por compresión y atrofia, pesan hasta 4kg cada uno. El epitelio de los quistes es plano y contienen un líquido seroso hemorrágico. Los quistes se ven desde la superficie externa.
La falla está en los genes de la Policistina 1 (85% de los casos) o 2 (15% de los casos), (PKD1 Y PKD2) complejos de proteinas de membrana del epitelio de los túbulos que regula la secreción de los mismos, en este caso la secreción de líquido es excesiva lo que provoca la dilatación de los túbulos formando quistes, primero microscópicos, que se presentan en la infancia, pero en edad adulta son macroscópicos y llevan a la insuficiencia renal alrededor de los 50 años. Estas proteinas participan de la interacción célula-célula y célula-matriz, interacciones necesarias para regular la proliferación y maduración del epitelio. Puede haber quistes en otros sectores: hígado, bazo, páncreas. La mutación PKD2 tarda mas en llegae a IRC.
-del niño: semejante a la anterior, pero lleva a la insuficiencia en la infancia, y los quistes macroscópicos no se ven desde la superficie externa la cual es lisa. La falla está en el gen de la Fibrocistina (PKHD1), proteina de membrana, sería un receptor necesario para la diferenciación de los túbulos colectores y de los conductos biliares.
* Enfermedades quisticas de la médula renal:
*Riñon en esponja medular: dilatación quistica de los colectores medulares. Etiología desconocida, se manifiesta en adultos, no trae consecuencias.
*Nefronoptisis/enfermedad quistica medular urémica: enfermedad renal que comienza en la infancia y puede llevar a la insuficiencia renal en edad adulta. Hay quistes medulares con atrofia cortical tubular y daño tubulointersticial, se conservan los glomérulos.
*Quistes adquiridos asociados a diálisis: quistes en corteza y médula, con epitelio plano, contenido seroso y cristales de oxalato de calcio. Se formarían por obstrucción de los túbulos (fibrosis o calcio), pueden sangrar. Se puede originar un carcinoma a partir del epitelio de los quistes.
*Quistes simples: únicos o múltiples, de 1 a 5 cm, contiene líquido claro, epitelio plano en única capa y se rodean por fuera de fibrosis. Se ubican en corteza, no dan síntomas, no confundirlos con tumores.



Glomerulonefritis
Procesos inflamatorios que afectan a los glomérulos y pueden llevar o no a la disfunción de los mismos.
Síndromes glomerulares:
1)Sme Nefrítico: hipertensión, hematuria macroscópica, azotemia, proteinuria y edemas leves a moderados.
2)Sme de GNF rapidamente progresiva: hematuria, proteinuria y oliguria grave que lleva a IRA en semanas.
3)Sme Nefrótico proteinuria mayor a 3,5 g por dia, hipoalbuminemia, edemas, hiperlipidemias, lipiduria, predisposicion a infecciones por pérdida de anticuerpos y factores del complemento, y es un estado de hipercoagulabilidad por pérdida de anticoagulantes por orina.
4)Sme de IRC: es la claudicación de un riñon insuficiente luego de un largo período de enfermedad. Comienza con azotemia (aumento de creatinina y de nitrógeno ureico en sangre), por un descenso del filtrado glomerular.
Alteraciones Histológicas:
1) Proliferación celular: de cèlulas mesangiales, endoteliales y parietales de la cápsula de Bowman.
2)Infiltrado leucocitario: neutrófilos, monocitos y a veces linfocitos.
3)Engrosamiento de Membrana basal glomerular: generalmente por depósito de complejos inmunes.
4)Esclerosis o hialinización: exceso de matriz mesangial y escape de proteínas plasmáticas. Ambas irreversibles.
Patogenia--------> Mecanismos Inmunológicos
1)Por Ac: _ por formación de complejos inmunes in situ (hipersens II)
_ por depósito de complejos inmunes (hipers III)
2)Por células: Linfocitos T
3)Por vía alterna del complemento: (C3)
Mecanismos de progresión del daño glomerular
1)Pèrdida del polianión glomerular: proteinuria
2)Hiperfiltración y cambios hemodinámicos
3)Fibrosis tubulointersticial
GLOMERULONEFRITIS PROLIFERATIVA AGUDA O POSTESTREPTOCÓCCICA
Es mas frecuente en niños y se da de 1 a 4 semanas deespués de una faringitis por EGA.
A veces puede darse por infección en la piel por el mismo streptococo y de GN.
Patogenia: mediada por Ac tipo III, los complejos de Ac y Ag ega, caen en glomérulos y dañan.
Histología: _ proliferación celular de c mesangiales, endotelio y rara vez, del epitelio parietal
_infiltrado leucocitario, neutrófilos y monocitos
_engrosamiento de la mb glomerular, debido a los complejos inmunes que están a nivel subepitelial y tienen forma de gibas.
Cambios difusos y globales.: en todos los glomérulos y en cada uno entero. Con Sme Nefrítico.
Evoluión: en el 95% de los casos se resuelve en 4-6 semanas. En el 4% pasa a la cronicidad y en el 1% pasan a rápidamente progresiva.


GLOMERULONEFRITIS RÁPIDAMENTE PROGRESIVA
Tipo I: *Sme GoodPasture: enfermedad autoinmune, se producen Ac contra colágeno tipo IV, cadena alfa3 de los tabiques alveolares y de la mb glomerular.
*Idiopática: ataque por Ac de los glomérulos, no se sabe contra qué Ag se dirigen.
Tipo II:*Postinfecciosa: son el 1% de las postestreptococcicas que evolucionan mal.
* Idiopática: deposita complejos inmunes, pero no se sabe el origen de los mismos.
Tipo III:*Granulomatosis de Wegener: presencia de C-ANCA
*Idiopática: presencia de C-ANCA y P-ANCA
Histología: Proliferación del epitelio parietal de Bowman, lo que forma semilunas. Se tapa la luz del espacio de Bowman. La fibrina estimula la proliferación del epitelio.


GLOMERULONEFRITIS MEMBRANOSA
Es la GN 1ria que cursa con Sme Nefrótico mas frecuente del adulto.
En el 85% de los casos se producen Ac cotra Ag renal aun no identificados, relacionado con HLA. En el 15%, es un Ag conocido que se ha pegado al riñon. El Ac ataca al Ag y al glomérulo.
Los Ag pueden deberse a infeccioes, sífilis, hepatitis B, C, paludismo. Por fármacos, AINES. Autoinmunes, lupus, tiroiditis.
Histología: engrosamiento de la mb glomerular, formación subepitelial pequeña y redondeada. Las luces vasculares se hallan disminuídas.

NEFROSIS LIPOIDEA/ ENFERMEDAD DE LOS CAMBIOS MÍNIMOS
Es la GN 1ria que cursa con Sme nefróticomas frecuente en niños, edad de 2 a 6 años.
Histología: glomérulos normales al MO, sólo presencia de lípidos dentro del epitelio tubular por lipiduria. Al Me se ve la pérdida difusa de los pedicelos. Es reversible porque los pedicelos regeneran.
Patogenia: Alteracion de linfocitos T, producen linfoquinas que serían tóxicas para el podocito.


GLOMERULOESCLEROSIS FOCAL Y SEGMENTARIA
Se puede dar tanto en niños como en adultos.
Patogenia: puede asociarse al HIV. Puede haber trastornos genéticos en las proteínas del podocito.
Hay lesión irreversible del podocito, muere y no se reemplaza.
Hay depósitos de complejos inmunes en el mesangio.
Histología: esclerosis focal y segmentaria que con el tiempo se hace difusa y global.


GLOMERULONEFRITIS MEMBRANOPROLIFERATIVA
Se da en niños y jóvenes.
Histología: engrosamiento de la mb glomerular con depósitos subendoteliales. Proliferación de c mesangiales, se interponen en la mb y la divide en dos: "en vía de tren".
Patogenia: tipo I: la mas frecuente, 66% de los casos, depósito de complejos.
tipo II: 34%, sólo depósito de complemento C3, activación de la via alterna.
Se ve al glomérulo como hiperlobulado, con las luces disminuídas.

GLOMERULONEFRITIS CRÓNICA
Los riñones están contraídos en forma simétrica, la corteza está adelgazada. En la mayoria de los casos se asocia a hipertension sistémica.
Histología: infiltrado leucocitario de mononucleares. Esclerosis global y difusa. Todos los glomérulos obliterados. Hialinización.


NEFROPATIA POR IGA O ENFERMEDAD DE BERGER
Se da en niños y jóvenes (la mas frecuente del mundo), al cursarla con hematuria no sintomática, habría muchos casos sin diagnosticar.
Patogenia: alteración genética por la cual se produce IgA en exceso ante estímulos como por ejemplo infecciones respiratorias altas. La IgA atrapa Ag y el complejo inmune se deposita en el sector del mesangio. Cuando se deposita activa al Co por via alterna.
Histología: proliferación de matriz y c mesangiales.
Asociada a enfermedad celíaca.

GN HEREDITARIAS
Sindrome de Alport: ligada a X, presenta sordera, trastornos visuales (cataratas, alteración de la córnea) y afectación glomerular: glomeruloesclerosis difusa por proliferación de la matriz.
Patogenia: existe un defecto hereditario en la síntesis de colágeno tipo 4, no se sintetiza la cadena alfa 5.
Clínica: los síntomas
comienzan entre los 5 y los 20 años de edad con hematuria micro o macroscópica, y llega a IRC entre los 20 y los 50 años de edad.
comienzan entre los 5 y los 20 años de edad con hematuria micro o macroscópica, y llega a IRC entre los 20 y los 50 años de edad.

Enfermedad de la membrana fina: o Hematuria benigna Familiar: la mb glomerular es mas delgada que en las personas normales, al ser mas fragil puede producir hematuria asintomática. La función renal se conserva normal. El defecto está en los genes que codifican las cadenas alfa 3 o alfa 4 del colágeno tipo 4.
Efermedades tubulares
Necrosis tubular aguda
Principal causa de IRA, 50% de todos los casos. Puede ser reversible. Se divide en:
_Isquémica: por circunstancias de shock, sepsis, quemaduras graves, hemólisis masiva (por hemoglobinuria), lesiones del músculo esquelético (mioglobinuria), etc.
Morfología: necrosis tubular focal en todos los niveles de la nefrona, separados por zonas indemnes. Hay rotura de las membranas basales (tubulorrexis) y oclusión de los túbulos distales por cilindros, son las proteínas de TAMM-HORSFALL, glucoproteína que normalmente secretan los túbulos.
_Nefrotóxica: por ingestión, inyección o inhalación de agentes tóxicos para los túbulos renales, como metales pesados (oro, plomo, mercurio), CCL4, alcohol metílico, ATB, pesticidas, anestésicos.
Morfología: hay necrosis en los túbulos proximales en forma continua. Se respetan las membranas basales y aparecen los cilindros proteicos en los túbulos distales.
Patogenia: a causa de isquemia o el tóxico, se produce liberación de renina, o de endotelinas o una disminución en la producción de NO, genera vasoconstricción preglomerular, disminuyendo el filtrado. Las células muertas y los cilindros obstruyen los túbulos.
Clínica:
1)Fase de comienzo: primeras 36hs después del tóxico o la isquemia
2)Fase oligúrica: la excreción de orina es de 50 a 400ml/dia, por el menor filtrado.
3)Fase diurética precoz: se reanuda el filtrado, pero como aun no hay capacidad de concentrar la orina puede deshidratarse y sufrir hipopotasemia que altere los músculos.
4)Fase de recuperación: se recupera la capacidad de concentrar y se normaliza, si esto ocurre, es después de la tercera semana.
Mejor sobrevida nefrotóxica (95%), si es que el tóxico no comprometió otro órgano; la isquémica tiene una sobrevida del 50%.

Enfermedades tubulointersticiales
Pielonefritis: infección renal que afecta a cálices, pelvis, túbulos e intersticio.
Los agentes son las bacterias de la flora fecal endógena: Escherichia Coli 50%, Proteus, Klebsiella, Enterobacter, Enterococo. Ortros, poliomavirus.
Vías de infección: -hemática: rara, por endocarditis, bacteriemias.
-ascendente: las bacterias suben desde la vejiga, primero se adhieren a la uretra, llegan a vejiga, se multiplican y por reflujo vesicoureteral llegan al riñon y por reflujo intrarrenal alcanzan las papilas.
Pielonefritis Aguda: hay supuración en intersticio y luego progresa hacia los túbulos provocando abscesos. Se observan neutrófilos. Los glomérulos se respetan, salvo que la infección sea micótica. Si la causa es el reflujo, el daño aparece en los polos. Complicaciones:
1)Necrosis papilar: mas frecuente en DBT, las papilas sufren necrosis de coagulación, sería por isquemia provocada por el edema inflamatorio. Si es bilateral lleva a IRA.
2)Pionefrosis: se produce cuando hay obstrucción de la vía urinaria, el exudado purulento se acumula en la vía y el riñon es un saco de pus.
3)Abscesos perirrenales: se extiende la supuración a los tejidos adyacentes
Pielonefritis crónica: Se produce por agudas repetidas, que llevan a la cicatrización de cálices y pelvis en forma irregular con daño tubulointersticial: los túbulos pueden estar atrofiados, hipertrofiados o dilatados. Hay infiltrado inflamatorio crónico, los glomérulos son los últimos en afectarse. 2 TIPOS:
1) por reflujo: la mas frecuente, eldaño está en los polos, por reflujo vesicoureteral.
2)por obstrucción: por anomalías congénitas, inflamaciones y tumores de la via urinaria, hiperplasia de próstata, cálculos. Al obstruir genera estasis.


TUMORES RENALES
Benignos:
*Adenoma papilar renal:
Se origina en el epitelio de los túbulos renales y forma papilas. Están siempre en la corteza, son amarillos, la cápsula es incompleta y miden no masde 0,5cm. Tienen la misma histologia que el hipernefroma y no se los puede diferenciar desde el punto de vista histologico ni por pruebas inmunohistoquímicas. Si es menos de 3 cm, sería adenoma, si mide mas, hipernefroma.

*Angiomiolipoma:
Formado por vasos, músculo liso y grasa. Se caracteriza por dar hematuria.


*Oncocitoma:
formado por células eosinofílicas, con núcleos pequeños, y están cargadas de mitocondrias. Se origina en las c intercaladas de los túbulos colectores. Está encapsulado y es de color bronceado. En raros casos metastatiza.
*Tumor de células yuxtaglomerulares:
Muy raro. Produce hipertensión secundaria por liberación de renina.
Malignos:
*Hipernefroma:
Neoplasia renal mas frecuente en los adultos (90%). Mas frecuente en varones de 60-70 años.
Color amarillo intenso. Nace en el epitelio tubular secretor renal, es un adenocarcinoma.
Morfología: mas frecuente en los polos, sobre todo en el superior. Miden de 3 a 15cm de diámetro. Color amarillo intenso con zonas de necrosis y hemorragias.
Invade pelvis, cálices, cápsulas, tiene particular tendencia a invadir la vena renal, creciendo como un cordón serpenteante dentro de la vena cava y llega a la aurícula derecha.
Metastatiza a ganglios regionales y por vía hemática: hígado, pulmón, hueso, cerebro.
Histología: pueden formar 4 patrones: papilar - sólido - tubular - trabecular
Tipos citológicos:
-Células claras: 80% de todos. Redondos o poligonales, citoplasma claro por contener glucógeno y lípidos.
-Células granulares: c eosinófilas cúbicas
-Células cromófobas: apenas eosinófilas, con un halo blanco perinuclear.
-Célula en tachuela: fusiformes.


Clínica:
tríada de Guyón: - dolor costovertebral
-masa palpable
- hematuria, es el elemento mas constante.
Smes Paraneoplásicos:
*fiebre, produe PGE2
*policitemia, aumenta EPO
*hipercalcemia, aumenta PTH
*HT, aumenta renina
*feminización y ginecomastia, aumenta gonadotrofina
*sme Cushing, aumenta ACTH
*reacciones leucemoides
*amiloidosis
Sobrevida: sin mts, a los 5 años 70%
con mts, a los 5 años 20%

Tumor de Wilms
Neoplasia renal mas frecuente en niños. Entre 2 y 5 años.
Predisponentes: nefroblastomatosis: focos de parénquima renal inmaduro, bilaterales. Smes genéticos con falla en cromosoma11.
Morfología: produce grandes masas esféricas que se observan a simple inspección, puede ser bilateral e el 5-10% de los casos.
Histología: tríada - blastema renal---> c pequeñas redondas y azules, embrionarias
- estroma renal----> músciulo liso, estríado, cartílago, hueso
- epitelio renal-----> glomérulos y túbulos abortivos
El pronóstico depende del grado de anaplasia de estos elementos.
Clínica: la madre consuolta por una masa visible y palpable.
Puede haber hematuria y dolor. Puede hacer mts a pulmón.